Aspartic
proteases
In
the last two decades the important biological role of aspartic proteases has
come into clear focus. In particular, it is now clear that enzymes of this class
are involved in several severe pathologies such as, for example, AIDS, cancer,
Alzheimer's disease, malaria.
|
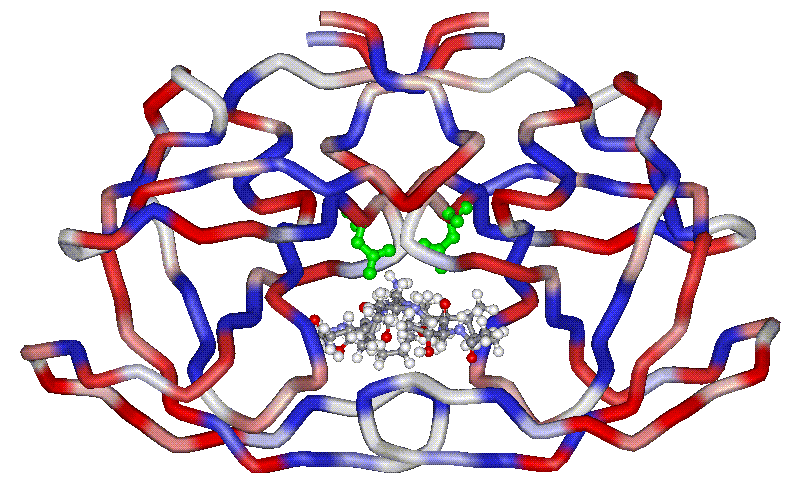
The aspartic protease from
HIV is a symmetric homodimer, formed by two monomers related by a C2
symmetry axis. The picture shows the crystal structure of HIV1-PR
complexed with an inhibitor. Carbon chains are coloured by hydrophobicity
of the residues.
|
Click on the picture to enlarge it |
The general acid-general base
mechanism, that is considered most likely for polypeptide hydrolysis catalyzed
by aspartic proteases (Figure), requires one Asp residue to be protonated, while
the second must be ionized and there is indeed experimental evidence that the
two aspartates share one proton at a physiological pH. The scissile amide bond
undergoes nucleophilic attack by a water molecule which is activated by the
deprotonated catalytic aspartic acid residue. The protonated aspartic acid
donates a proton to the amide bond nitrogen, generating a tetrahedral
zwitterionic intermediate which collapses to the cleaved products with a similar
mechanism, in the slow step of the reaction.[i]
An additional water molecule binds between the substrate and the main chain
amide groups of the enzyme (at the level of Ile50 and Ile50’) and is thought
to twist the scissile peptide bond out of planarity, thereby facilitating the
cleavage of this bond.[ii]
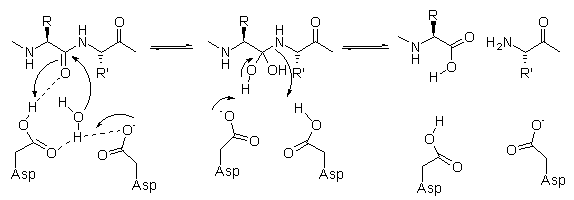
Mechanism of hydrolysis of
aspartic proteases.
|
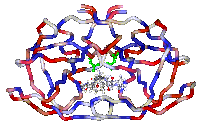
The active site of aspartic
proteases does not in general contain groups that are sufficiently nucleophilic
to be chemically modified by a selective irreversible inhibitor. The catalytic
aspartates can be alkylated only by strong electrophiles, such as epoxides, that
are potentially cytotoxic.[iii]
The picture shows the active site with the two catalytic aspartates
coloured green.
|

Click on the picture to
enlarge it. |
Thus, most aspartic protease
inhibitors that have been developed to date bind to their target enzyme through
noncovalent interactions (i.e. hydrogen bonds, ionic or van der Waal’s
contacts). These compounds are therefore reversible inhibitors, and an effective
inhibition results when the enzyme has higher affinity for the inhibitor than
for its natural substrate. It has been proposed that stable structures which
resemble the transition state of an enzyme catalyzed reaction should bind the
enzyme more tightly than the substrate. An approach that has proved very
successful for the design of efficient aspartic protease inhibitors is based on
the incorporation of a transition-state analog into a peptidomimetic structure.
A transition-state isostere is defined as a functional group that can mimic the
tetrahedral transition-state of amide bond hydrolysis, but is stable and non
hydrolyzable.
|
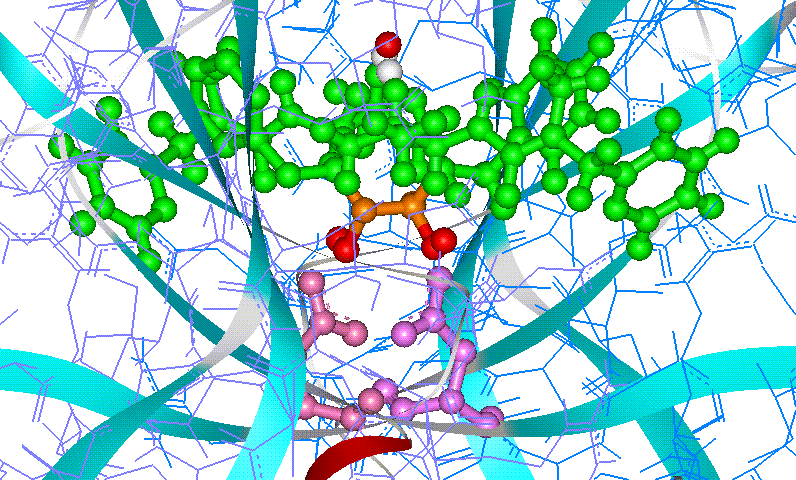
Among transition-state isosteres that have been successfully used
in the design of aspartic protease inhibitors, hydroxy-based isosteres have
proved particularly effective.
The picture shows how inhibitor A76889 fits into the active site of
HIV1-PR. |

Click on the picture to
enlarge it. |
Crystallographic
structures of aspartic proteases can be downloaded from the Protein
Data Bank. We used Web Lab Viewer to render the images.
[i]
Hyland, L. J.; Tomaszek, T. A.; Meek, T. D. Biochemistry 1991, 30,
8454-8463.
[ii]
Chatfield, D. C.; Brooks, B. R. J. Am.
Chem. Soc. 1995, 117,
5561-5572.
[iii]
a) Salto, R.; Babè, L. M.; Li, J.; Rosè, J. R.; Yu, Z.; Burlingame, A.;
DeVoss, J. J.; Sui, Z.; Ortiz de Montellano, P.; Craick, C. S. J.
Mol.Biol. 1994, 269, 10691; b) Abell,
A. D.; Hoult, D. A.; Bergman, D. A.; Fairlie, D.P.Bioorg. Med. Chem.
Lett. 1997, 7, 2853; c) DeVoss, J. J.; Sui, Z.; DeCamp, D. L.; Salto, R.; Babè,
L. M.; Craick, C. S.; Ortiz de Montellano, P. J. Med. Chem. 1994, 37, 665; d) Jones, P. R.; Burlingame, A. L.; Kuntz, I. D.; Craick,
C. S.; Ortiz de Montellano P. R. J. Am.
Chem.
Soc. 1996,
118, 5846; e) Said, B.; Matsumoto,
D. C.; Hamade, A. K.; Shank, R. C. Biochem.
Biophys.
Res. Commun. 1999,
261, 844-847.