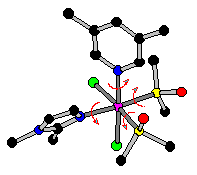
Molecular mechanics force field constants for ruthenium(II) sulfoxide complexes have been derived for the implementation of the Amber force field. Stretching and bending constants were calculated by the Badger's and Halgren's equations. Twenty-six parameters for sulfoxide, chlorine and metal atoms have been optimized to fit 836 experimental bond lengths and angles, using the Simplex method. Atomic charges have been calculated by the semi-empirical method ZINDO/1. The goal was to rationalize the stereochemical and conformational features of this kind of metal complexes, through an accurate description of the molecular structures. The accuracy in the resulting geometries is reflected in the low values of the average differences between observed and calculated bond lengths and angles (-0.007 Å and 0.0 °) and their root-mean-square-deviations (0.017 Å and 1.3 °). Conformational analyses for S- and O- bonded sulfoxides have been carried out. Possible rotamer distributions for ruthenium(II)-dimethyl sulfoxide complexes containing lopsided nitrogen ligands, arising from hindered rotation about the Ru-N bonds, also have been investigated.
![]() Structural Chemistry Group Home Page
Structural Chemistry Group Home Page